An R package for weighted region comethylation network analysis.
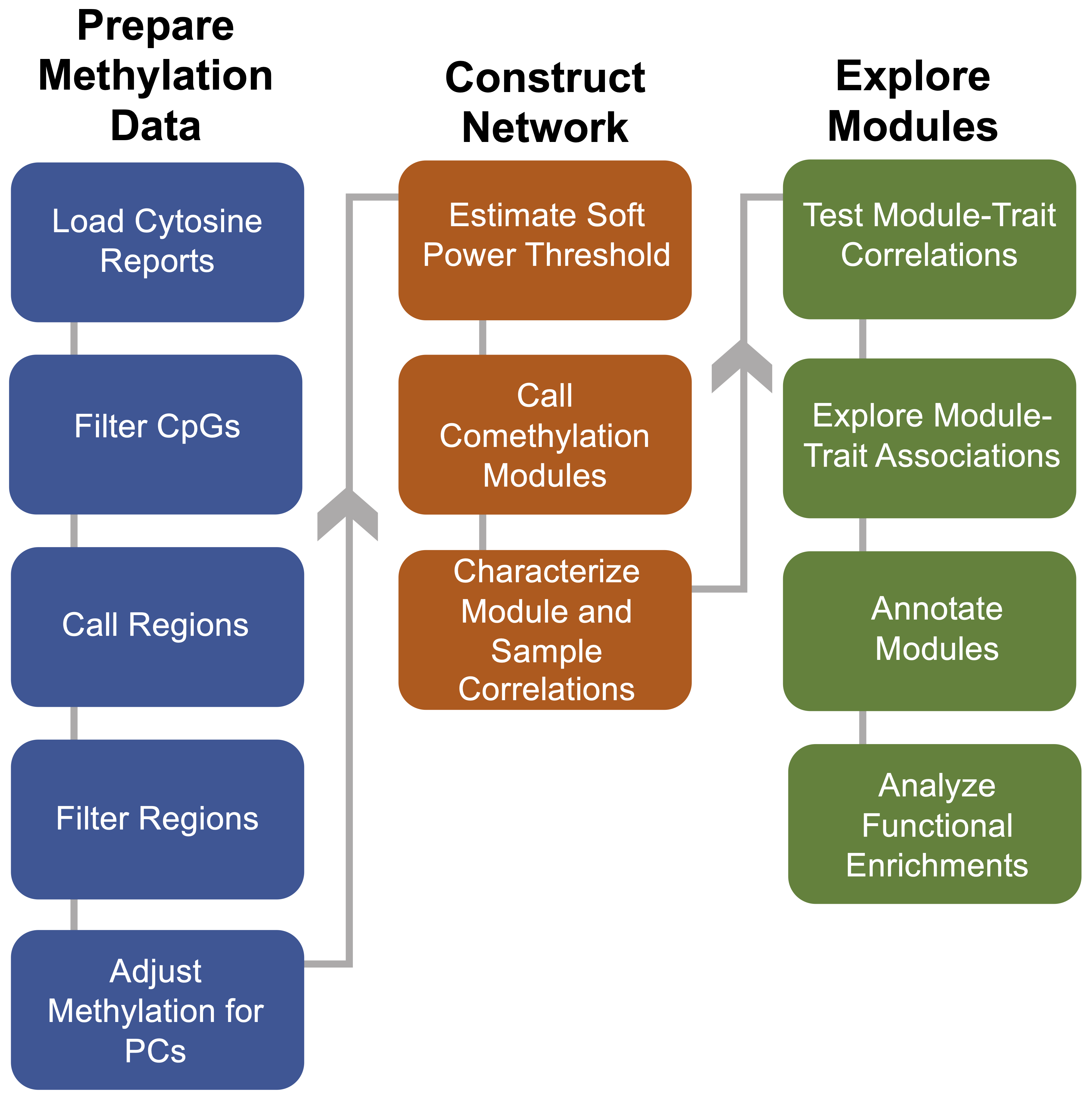
Comethyl builds upon the WGCNA package to identify and interpret modules of comethylated regions from whole-genome bisulfite sequencing data. Regions are defined from clusters of CpG sites or from genomic annotations, and then percent methylation values are used to identify comethylation modules.
Interesting modules are identified and explored by comparing with sample traits and examining functional enrichments. Results are then visualized with high-quality, editable plots from ggplot2.
Installation
You can install Comethyl from this repository and load it into your R session with the code below.
install.packages(c("BiocManager", "remotes"))
BiocManager::install("cemordaunt/comethyl")
library(comethyl)Documentation
Complete documentation for comethyl is available at https://cemordaunt.github.io/comethyl/.
Citing Comethyl
If you use Comethyl in your work, please cite our Briefings in Bioinformatics publication:
Mordaunt CE, Mouat JS, Schmidt RJ, and LaSalle JM. (2022) Comethyl: a network-based methylome approach to investigate the multivariate nature of health and disease. Briefings in Bioinformatics bbab554.