Module Preservation
Charles Mordaunt
Source:vignettes/Module_Preservation.Rmd
Module_Preservation.RmdIntroduction
In this vignette, we use Comethyl to construct a comethylation network using clusters of CpGs grouped by genomic location in a replication dataset, and then examine the preservation of modules from the original dataset. We identify modules of comethylated regions, generate statistics of module preservation, and visualize them.
The original dataset explored in the CpG Cluster Analysis vignette included 74 male cord blood samples from newborns who were later diagnosed with autism spectrum disorder (ASD) and those with typical development (TD). The replication dataset has the same design and includes 38 male cord blood samples. The goal of this analysis is to evaluate the consistency of our identified modules in an independent group of individuals.
Raw data is available on GEO (GSE140730), see the previous publication for more details.
Set Global Options
WGCNA::disableWGCNAthreads() prevents multi-threading for WGCNA calculations, including all functions using WGCNA::cor() and WGCNA::bicor(). This is recommended for large sets of regions (> 150,000). For smaller region sets, use WGCNA::enableWGCNAthreads() to allow for parallel calculations with the specified number of threads. If the number of threads is not provided, the default is the number of processors online.
options(stringsAsFactors = FALSE)
Sys.setenv(R_THREADS = 1)
WGCNA::disableWGCNAThreads()Read Bismark CpG Reports
In this first section, we try to match the CpGs and regions from the analysis of the first dataset to make them more comparable.
We read in an excel table with openxlsx::read.xlsx() where the first column includes the names of sample Bismark CpG reports and all other columns include trait values for each sample. All trait values must be numeric, though traits can be categorical or continuous. getCpGs() reads individual sample Bismark CpG reports into a single BSseq object and then saves it as a .rds file. See Inputs for more information.
We load the filtered BSseq object from the first analysis and use IRanges::subsetByOverlaps() to limit our new BSseq object to those same CpGs. Then filterCpGs() subsets a BSseq object to include only those CpGs meeting cov and perSample cutoffs and then saves it as a .rds file. filterCpGs() is designed to be used after cov and perSample arguments have been optimized by getCpGtotals() and plotCpGtotals(). Here we keep only CpGs with at least 1 read in at least 1 sample. We make this more inclusive than usual to better match the original dataset.
colData_rep <- openxlsx::read.xlsx("replication_sample_info.xlsx", rowNames = TRUE)
bs_rep <- getCpGs(colData_rep, file = "Replication_Unfiltered_BSseq.rds")
bs_disc <- readRDS("Discovery_Filtered_BSseq.rds")
bs_rep <- IRanges::subsetByOverlaps(bs_rep, ranges = bs_disc)
bs_rep <- filterCpGs(bs_rep, cov = 1, perSample = 0.01, file = "Replication_Filtered_BSseq.rds")Call Regions
We read in the regions from the original dataset and create a GRanges object. getRegions() generates a set of regions and some statistics based on the CpGs in a BSseq object. Here we use the original regions as a custom genomic annotation and input into getRegions(). It’s also necessary to replace RegionID with the name column so that the RegionID’s will match between the two datasets.
Next we plot the region statistics. plotRegionStats() plots histograms of region statistics, while plotSDstats() plots methylation standard deviation versus region statistics. With these plots, we can get an idea of the characteristics of our regions and see how methylation variability is affected. The goal is to identify regions with biological variability rather than technical variability (due to low coverage).
regions_disc <- read.delim("Discovery_Filtered_Regions.txt") %>%
select(chr, start, end, RegionID)
regions_disc <- with(regions_disc, GenomicRanges::GRanges(seqnames = chr, ranges = IRanges::IRanges(start = start, end = end), name = RegionID))
regions_rep <- getRegions(bs_rep, custom = regions_disc, n = 1, save = FALSE) %>%
select(RegionID = name, chr:methSD)
write_tsv(regions_rep, file = "Replication_Unfiltered_Regions.txt")
plotRegionStats(regions_rep, maxQuantile = 0.99, file = "Replication_Unfiltered_Region_Plots.pdf")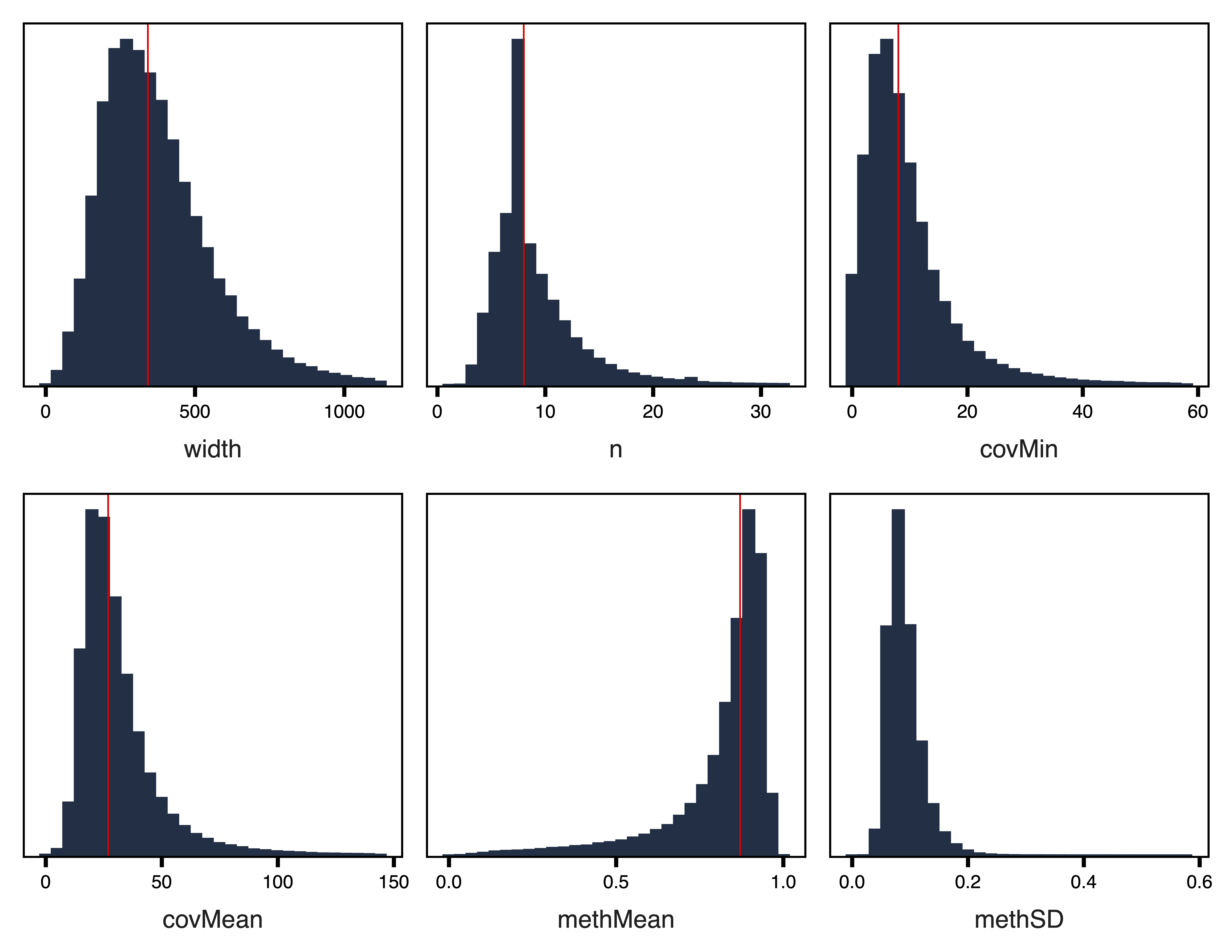
Figure 1. Unfiltered Region Plots
plotSDstats(regions_rep, maxQuantile = 0.99, file = "Replication_Unfiltered_SD_Plots.pdf")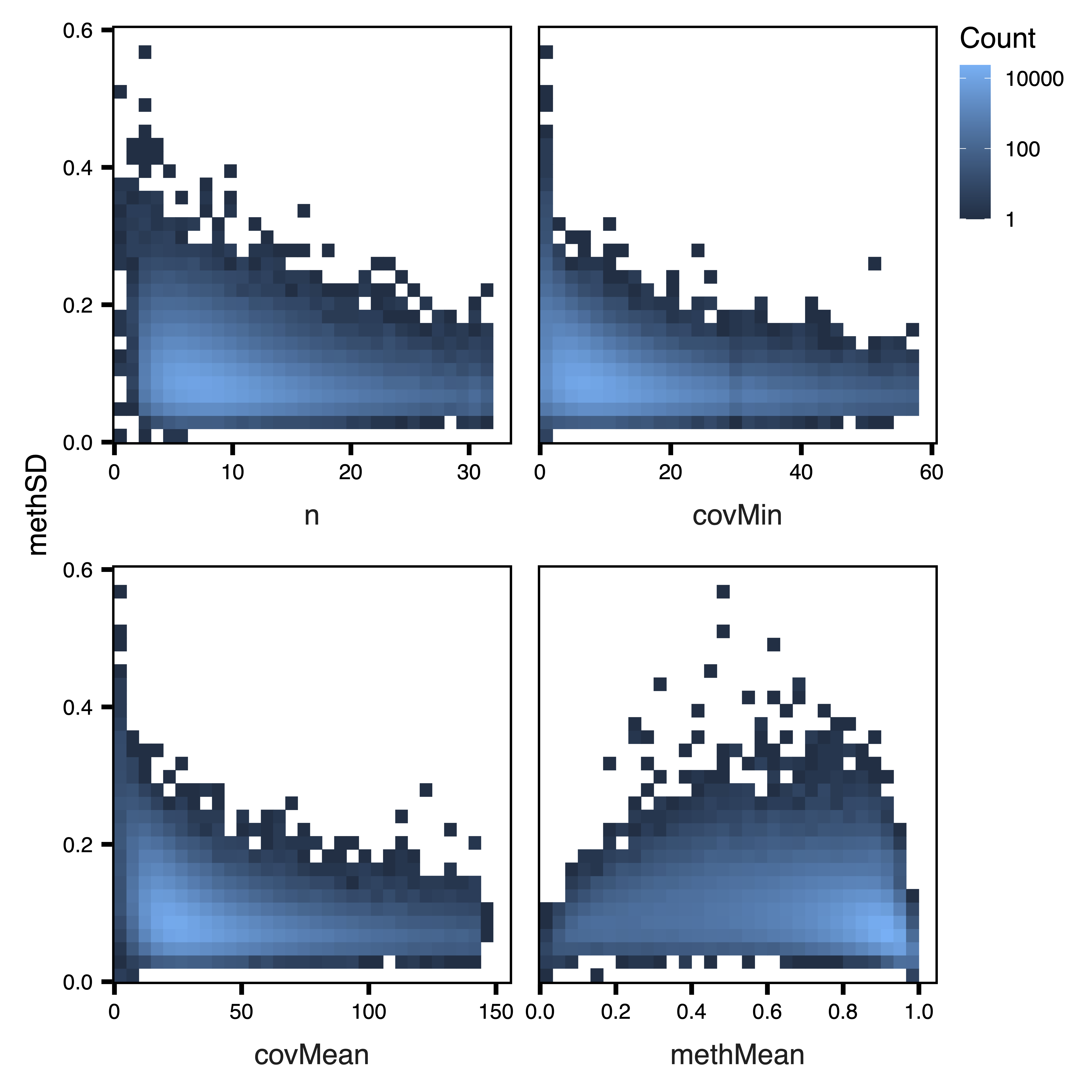
Figure 2. Unfiltered SD Plots
Examine Region Totals at Different Cutoffs
getRegionTotals() calculates region totals at specified covMin and methSD cutoffs. Total regions (and thus total width and CpGs) are expected to decrease as the minimum coverage cutoff increases and SD cutoff increases. plotRegionTotals() plots these region totals by potential covMin and methSD cutoffs.
regionTotals_rep <- getRegionTotals(regions_rep, file = "Replication_Region_Totals.txt")
plotRegionTotals(regionTotals_rep, file = "Replication_Region_Totals.pdf")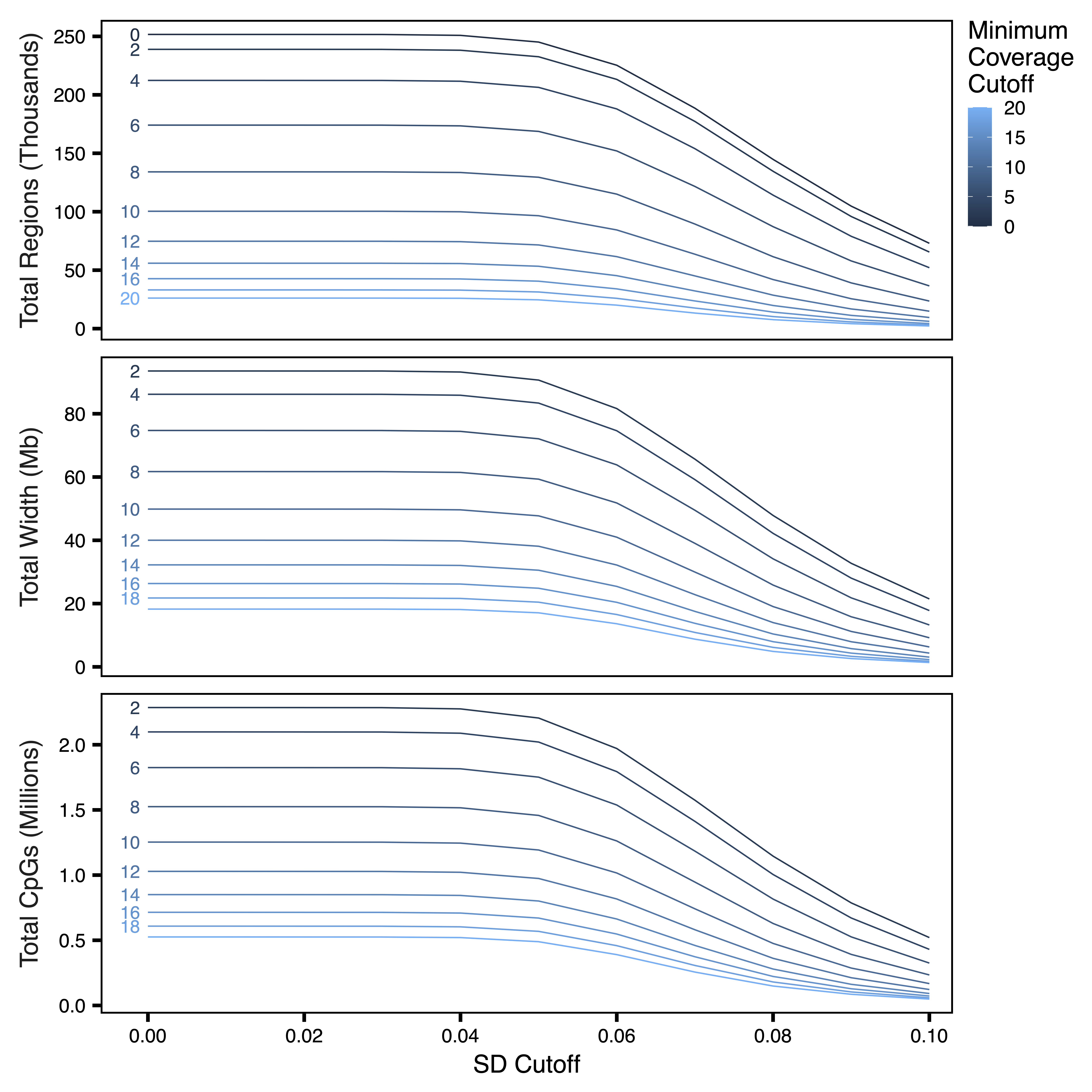
Figure 3. Region Totals
Filter Regions
filterRegions() subsets the regions to only include those meeting covMin and methSD cutoffs. filterRegions() is designed to be used after covMin and methSD functions have been optimized with getRegionTotals() and plotRegionTotals(). Here we filter for regions with at least 5 reads in all samples and with a methylation standard deviation of at least 5%. Then we examine our regions again with plotRegionStats() after filtering and continue with extracting methylation data and constructing our network.
regions <- filterRegions(regions_rep, covMin = 5, methSD = 0.05, file = "Replication_Filtered_Regions.txt")
plotRegionStats(regions_rep, maxQuantile = 0.99, file = "Replication_Filtered_Region_Plots.pdf")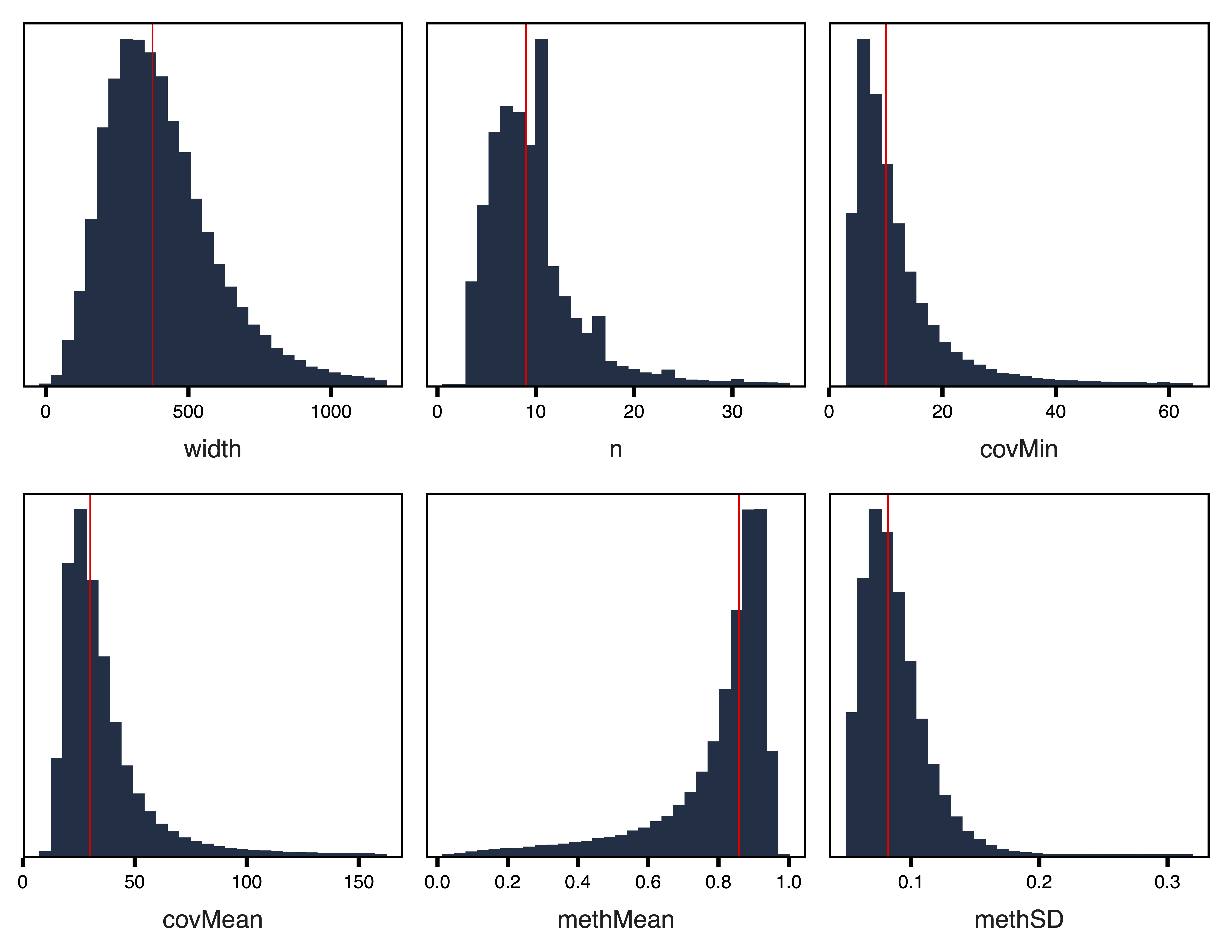
Figure 4. Filtered Region Plots
Adjust Methylation Data for Principal Components
getRegionMeth() calculates region methylation from a BSseq object and saves it as a .rds. model.matrix() creates a design matrix for our set of samples. getPCs() calculates the top principal components, and then adjustRegionMeth() adjusts the region methylation for the top PCs and saves it as a .rds file. getDendro() clusters the samples based on the adjusted region methylation using Euclidean distance, while plotDendro() plots the dendrogram. We can use this dendrogram to see if there are any outlier samples or samples clustering separately due to batch effects.
meth_rep <- getRegionMeth(regions_rep, bs = bs_rep, file = "Replication_Region_Methylation.rds")
mod_rep <- model.matrix(~1, data = bsseq::pData(bs_rep))
PCs_rep <- getPCs(meth_rep, mod = mod_rep, file = "Replication_Top_Principal_Components.rds")
methAdj_rep <- adjustRegionMeth(meth_rep, PCs = PCs_rep, file = "Replication_Adjusted_Region_Methylation.rds")
getDendro(methAdj_rep, distance = "euclidean") %>%
plotDendro(file = "Replication_Sample_Dendrogram.pdf", expandY = c(0.25, 0.08))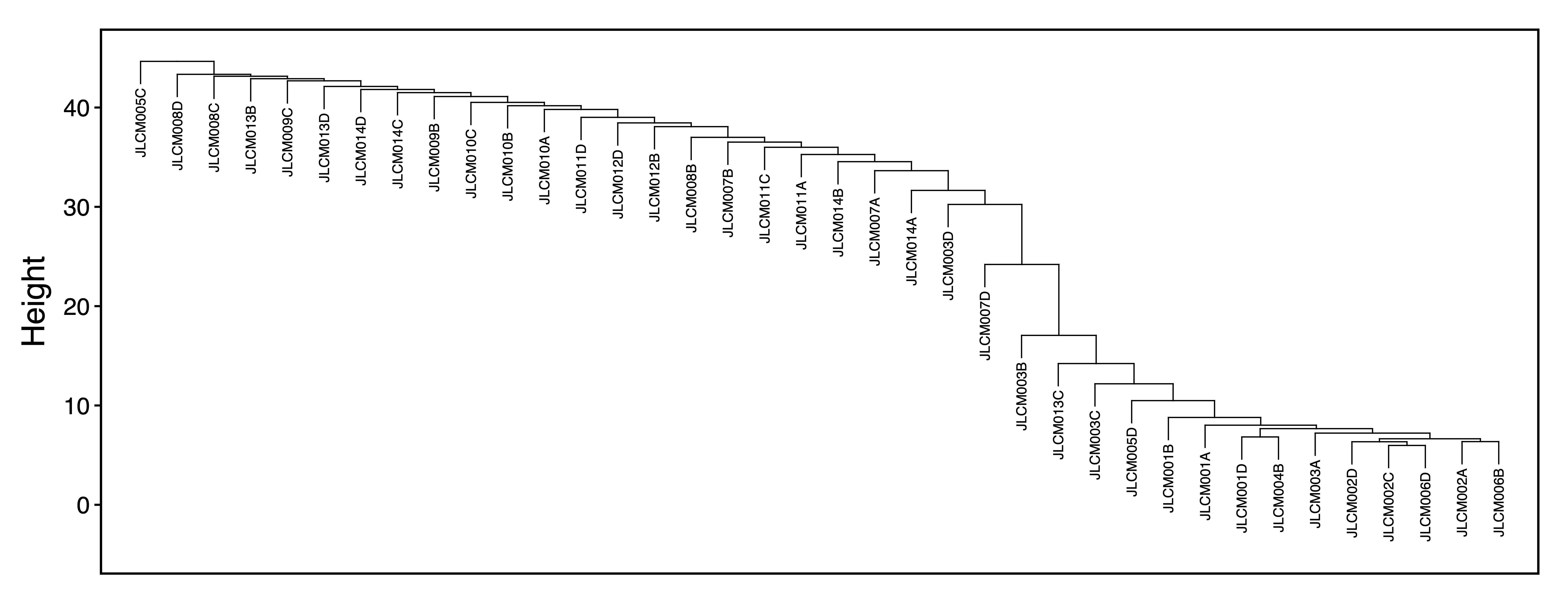
Figure 5. Sample Dendrogram
Select Soft Power Threshold
getSoftPower() analyzes scale-free topology with Pearson or Bicor correlations to determine the best soft-thresholding power. This refers to the power to which all correlations are raised and how much more stronger correlations are weighted compared to weaker correlations. Pearson correlation is more sensitive than Bicor correlation, but is also more influenced by outlier samples. We use Pearson correlation in order to have higher power to detect correlated regions in a dataset with relatively low variability between samples.
plotSoftPower() plots the soft power threshold against scale free topology fit and connectivity. Typically, as the soft power threshold increases, fit increases and connectivity decreases. A soft power threshold should be selected as the lowest threshold where fit is 0.8 or higher. After originally trying 19, we increase the power to 25 for better fit and a mean connectivity that more closely matches the first dataset.
sft_rep <- getSoftPower(methAdj_rep, powerVector = 1:30, corType = "pearson", file = "Replication_Soft_Power.rds")
plotSoftPower(sft_rep, file = "Replication_Soft_Power_Plots.pdf")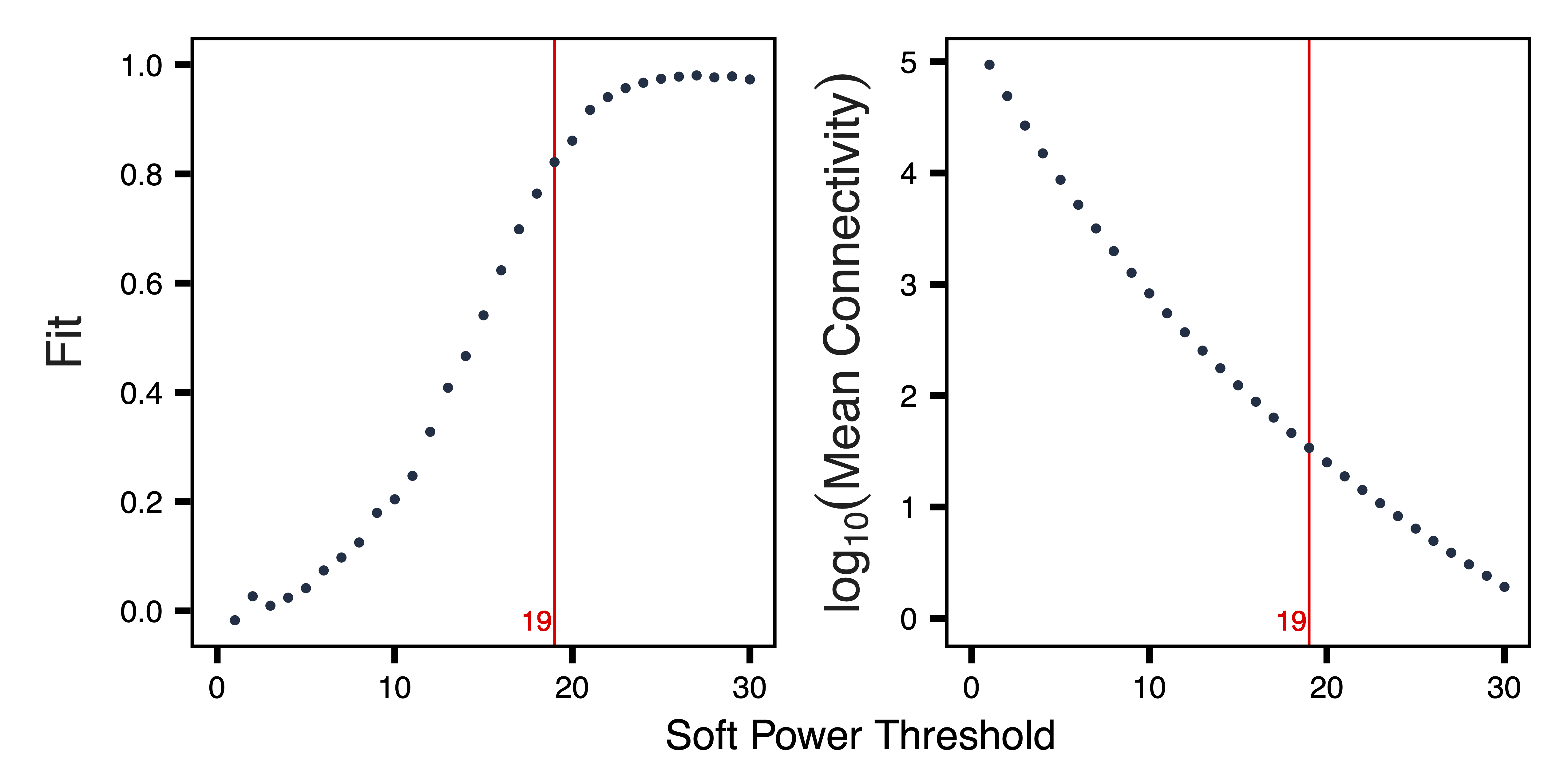
Figure 6. Soft Power Plots
Get Comethylation Modules
getModules() identifies comethylation modules using filtered regions, a chosen soft power threshold, and either Pearson or Bicor correlation. Here we use Pearson correlation for the greater sensitivity to detect modules. Regions are first formed into blocks close to but not exceeding the maximum block size. A full network analysis is then performed on each block to assign regions to modules; modules are merged if their eigennodes are highly correlated. The modules are then saved as a .rds file. This two-level clustering approach requires less computational memory and is significantly faster than performing network analysis on all regions at once.
modules_rep <- getModules(methAdj_rep, power = 25, regions = regions, corType = "pearson", nThreads = 2, file = "Replication_Modules.rds")Evaluate Module Preservation
After module identification and functional association has been completed within a dataset, an important next step is the analysis of module quality and preservation in an independent replication dataset. One approach is that taken by Langfelder et al., where multiple statistics are calculated to assess different types of preservation based on overlapping module assignments and network connectivity features. Observed statistics are then compared with an empirical null distribution, generated by permuting module assignments 100 times, in order to calculate Z-scores and P-values. In simulation studies by Langfelder et al., Z-scores > 2 were estimated as weak-to-moderate evidence for preservation, while Z-scores > 10 were estimated as strong evidence.
To test module preservation, we load the regions with module assignments and methylation data for the original dataset and input them into getModulePreservation() along with the new regions and associated methylation data. This calculates several preservation statistics for each module, along with Z-scores and p-values comparing them to the empirical null distribution. After waiting for the permutations to complete, we run plotModulePreservation() to visualize the preservation analysis. This function plots the Z-scores by module size, due to the correlation of some of the statistics with the number of regions in the module.
As given by the summary.qual Z-score, all modules were scored as having strong evidence for good quality, except for the Ivory module, which had weak/moderate evidence. Using the cross-tabulation derived accuracy Z-score, 40/53 modules had at least some evidence for preservation. In contrast, relatively few modules had evidence for preservation across these two datasets based on the network-derived summary.pres Z-score. Preserved modules with this statistic included the Light-Cyan, Blue and Floral-White modules. This example demonstrates the value of multiple measurements of module quality and preservation in identifying robust and reproducible comethylation modules.
modules_disc <- readRDS("Discovery_Modules.rds")
regions_disc <- modules_disc$regions
methAdj_disc <- readRDS("Discovery_Adjusted_Region_Methylation.rds")
regions_rep <- modules_rep$regions
preservation <- getModulePreservation(methAdj_disc, regions_disc = regions_disc, meth_rep = methAdj_rep, regions_rep = regions_rep, corType = "pearson", file = "Module_Preservation_Stats.txt")
plotModulePreservation(preservation, file = "Module_Preservation_Plots.pdf")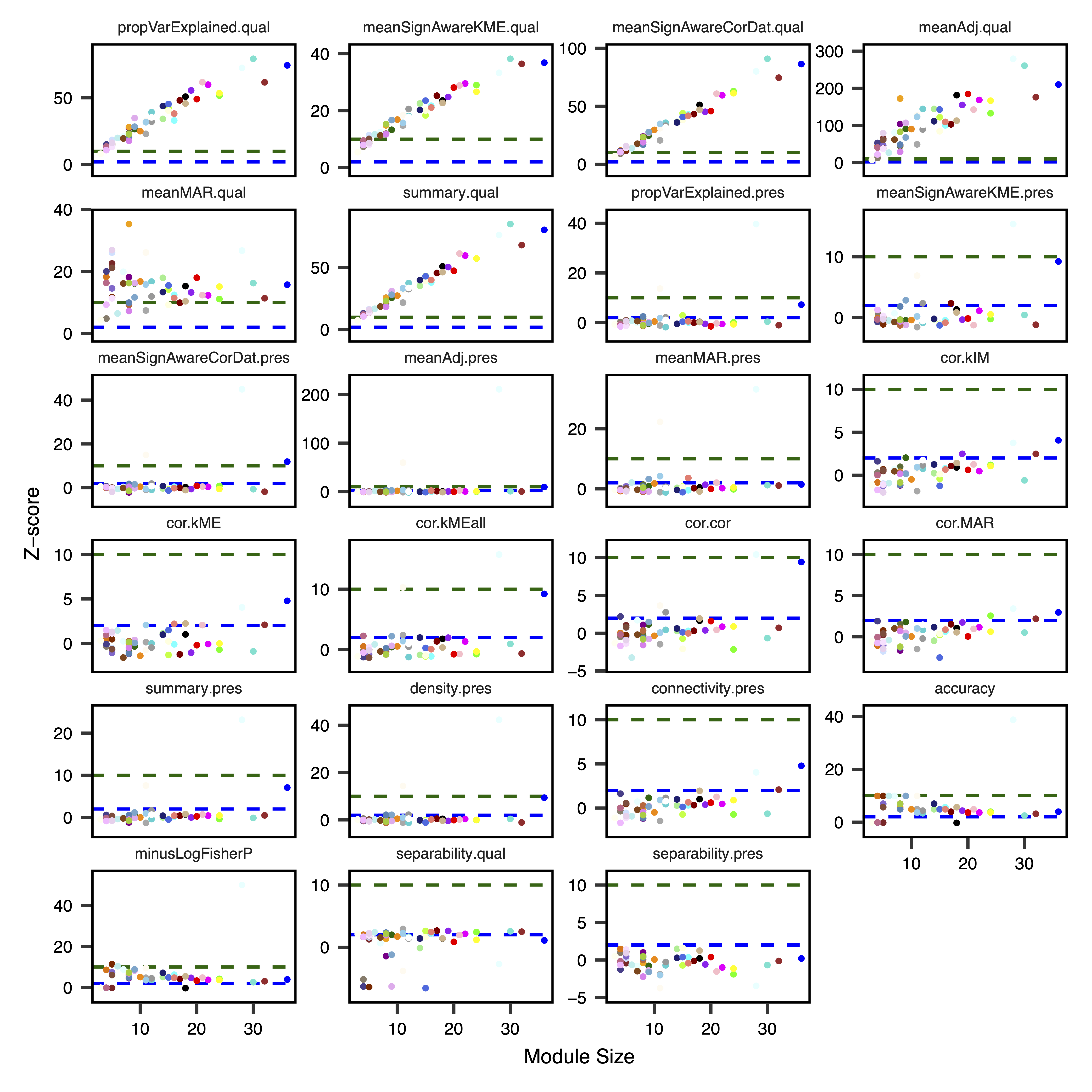
Figure 7. Module Preservation Statistic Plots